|
Stato Solido Capitoli:
Appendici |
TightBinding< Condizioni al contorno di Born-Von Karman e numero degli stati | Indice | Bande e classificazione dei solidi > Fino ad ora abbiamo discusso una classificazione degli stati elettronici in un cristallo basata su considerazioni puramente geometriche. Per studiare la dinamica degli elettroni occorre risolvere l'equazione di Schrödinger, della quale abbiamo sfruttato per adesso solo le proprietŕ di simmetria traslazionale. Ci aspettiamo di ricavare gli autovalori dell'energia (i livelli energetici) per ciascuno stato identificato da un valore di {$\mbox{\it\bf k}$} all'interno della prima zona di Brillouin del reticolo reciproco, ossia una funzione {$\varepsilon=\varepsilon(\mbox{\it\bf k})$}. Mostreremo che la distribuzione dei livelli energetici costituisce un quasi continuo di valori permessi, interrotto da intervalli di valori proibiti; questo andamento, descritto dalla funzione {$\varepsilon(\mbox{\it\bf k})$}, definisce la struttura a bande di energia del solido e l'esistenza di intervalli proibiti tra le bande č dovuta alla natura periodica del reticolo reciproco. Allo scopo di illustrare come insorge la struttura a bande, iniziamo dal cosiddetto legame forte, o tight binding, che considera gli elettroni ancora prevalentemente associati ciascuno al proprio atomo; si tratta quindi del modello naturale per gli elettroni piů esterni dei materiali non metallici (isolanti e semiconduttori), ma, come vedremo, esso č rilevante anche per i metalli di transizione. Questo modello, oltre a costituire un approccio realistico alla struttura elettronica di diversi materiali, consente di confrontare direttamente i livelli energetici del solido con quelli della specie atomica che lo compone, mettendo in luce il meccanismo di formazione delle bande. Supponiamo di avere a che fare con un solido monoatomico del quale siano note le funzioni d'onda dell'atomo isolato, {$\psi^a_n(\mbox{\it\bf r})$}, la cui densitŕ di probabilitŕ va rapidamente a zero al di lŕ di un certo raggio. {${\cal H}^a$} č l'hamiltoniana atomica e l'indice {$n$} rappresenta i numeri quantici dello stato (ad es. {$1s$}, {$2p$}, ...); si puň scrivere quindi {$ (27)\qquad\qquad {\cal H}^a\psi^a_n(\mbox{\it\bf r})=\{ -\frac {\hbar^2}{2m}\nabla^2+{\cal V}^a(\mbox{\it\bf r})\}\psi^a_n(\mbox{\it\bf r}) = \varepsilon^a_n\psi^a_n(\mbox{\it\bf r}) .$} Possiamo immaginare di costruire il solido avvicinando gradatamente tra di loro gli {$N$} atomi che lo compongono; questo assemblaggio ipotetico del solido mantiene ad ogni stadio la struttura periodica del cristallo reale, partendo da uno stato estremamente rarefatto. Se trattiamo gli elettroni con il campo autoconsistente ad ogni stadio intermedio la loro hamiltoniana conterrŕ un potenziale periodico della forma vista nell'equazione 2, nel quale, per il momento, trascuriamo il termine di scambio: {$ (28) \qquad\qquad {\cal H}=- \frac{\hbar^2} {2m}\nabla^2+{\cal V}(\mbox{\it\bf r}) $}. Se all'inizio gli atomi sono lontani tra di loro (il passo reticolare č molto grande rispetto al raggio atomico) si puň considerare il potenziale come somma dei potenziali atomici centrati sui diversi nodi {$\mbox{\it\bf R}$} del reticolo rarefatto, {$\qquad\qquad {\cal V}(\mbox{\it\bf r})=\sum_{\mbox{\it\bf R}}{\cal V}^a(\mbox{\it\bf r}+\R)$}. Ciň equivale a trascurare l'elemento di matrice del potenziale di un atomo sulle autofunzioni centrate su atomi diversi. Si verifica immediatamente che in queste condizioni ciascuna funzione atomica {$\psi^a_n(\mbox{\it\bf r}-\mbox{\it\bf R})$}, indipendentemente dal nodo {$\mbox{\it\bf R}$} su cui ha centro, č autostato di {$\cal H$} con il medesimo autovalore {$\varepsilon_n^a$} Per semplicitŕ ci concentreremo nel seguito su una funzione atomica non degenere di tipo {$s$}, (se considerassimo funzioni {$p,d,\cdots$} dovremmo sostituire alla singola {$\psi^a_n$} una opportuna combinazione lineare delle funzioni degeneri che corrispondono a questi livelli). Con le {$N$} funzioni atomiche, divenute degeneri nel cristallo, si puň costruire l'onda di Bloch di vettor d'onda {$\mbox{\it\bf k}$} {$ (29) \qquad\qquad \psi_{n\mbox{\it\bf k}}(\mbox{\it\bf r})=N^{-\frac 1 2} \sum_{\mbox{\it\bf R}} \e^{i\mbox{\it\bf k}\cdot\mbox{\it\bf R}} \psi^a_n(\mbox{\it\bf r}-\R). $} č immediato verificare che si tratta proprio di una funzione di Bloch; per mostrarlo basta calcolarne l'autovalore dell'operatore di traslazione, {$ \qquad\qquad{\cal T}_{\mbox{\it\bf R}^\prime}\psi_{n\mbox{\it\bf k}}=\e^{i\mbox{\it\bf k}\cdot\mbox{\it\bf R}^\prime}\psi_{n\mbox{\it\bf k}}$}, ridefinendo il vettore corrente {$\mbox{\it\bf R}$} della sommatoria della 29 come {$\mbox{\it\bf R} - \mbox{\it\bf R}^\prime$}. La funzione {$\psi_{n\mbox{\it\bf k}}$}, che nella sua versione piů completa č un equivalente della funzione MOLCAO? (nei solidi la si chiama semplicemente LCAO), ha su ciascun sito l'andamento tipico delle funzioni atomiche ed č modulata di sito in sito dall'onda piana, come č mostrato in figura 22.
Essa č anche normalizzata, finchč la sovrapposizione tra funzioni centrate su siti distinti č trascurabile. In quest'approssimazione, ossia finchč il potenziale dell'equazione 28 coincide nell'intorno di ogni atomo con il corrispondente potenziale atomico, {$\psi_{n\mbox{\it\bf k}}$} č un autostato dell'elettrone nel cristallo rarefatto. Se perň continuiamo ad avvicinare tra loro gli atomi la sovrapposizione tra le funzioni d'onda atomiche centrate su siti distinti non č piů trascurabile, e neppure lo č l'elemento di matrice del potenziale atomico sulla funzione d'onda del nodo vicino. Ciň significa che le funzioni d'onda iniziano a rimescolarsi tra loro e, di conseguenza, a produrre un differente potenziale autoconsistente. Il potenziale periodico cristallino non sarŕ piů la semplice somma dei potenziali atomici e dovremo incominciare a trattare {$\qquad\qquad \delta{\cal V}={\cal V} - \sum_{\mbox{\it\bf R}}{\cal V}^a(\mbox{\it\bf r}+\mbox{\it\bf R})$} come una perturbazione. Rispetto ad essa la funzione {$\psi_{n\mbox{\it\bf k}}$} dell'equazione 29 rappresenta l'autostato all'ordine zero, che serve per ricavare perturbativamente come si spostano i livelli energetici al primo ordine in questa graduale costruzione del solido; in particolare {$\varepsilon_n(\mbox{\it\bf k})$} varia di {$ (30)\qquad\qquad\varepsilon_n(\mbox{\it\bf k})-\varepsilon^a_n = \int \psi_{n\mbox{\it\bf k}}^*(\mbox{\it\bf r})\delta{\cal V}(\mbox{\it\bf r})\psi_{n\mbox{\it\bf k}}(\mbox{\it\bf r})d\mbox{\it\bf r} ,$} dove si č trascurata la correzione dovuta alla normalizzazione delle {$\psi_{n\mbox{\it\bf k}}$}, che contribuisce solo a ordini superiori in {$\delta{\cal V}$}. Sostituendo l'autostato all'ordine zero (equazione 29}) l'integrale si sviluppa in {$ (31)\qquad\qquad\begin{eqnarray}\varepsilon_n(\mbox{\it\bf k})-\varepsilon^a_n & = & N^{-1} \sum_{\mbox{\it\bf R},\mbox{\it\bf R}^\prime}\int \psi^{a*}_n(\mbox{\it\bf r}-\R)\delta{\cal V}(\mbox{\it\bf r})\psi^a_n(\mbox{\it\bf r}-\mbox{\it\bf R}^\prime)d\mbox{\it\bf r}\,\,e^{i\mbox{\it\bf k}\cdot(\mbox{\it\bf R}^\prime-\mbox{\it\bf R})} \\& = &\sum_{\mbox{\it\bf R},\mbox{\it\bf R}^\prime} \frac {e^{-i\mbox{\it\bf k}\cdot\mbox{\it\bf R}}} N \int \psi^{a*}_n(\mbox{\it\bf r}-\mbox{\it\bf R}-\mbox{\it\bf R}^\prime)\delta{\cal V}(\mbox{\it\bf r}-\mbox{\it\bf R}^\prime)\psi^a_n(\mbox{\it\bf r}-\mbox{\it\bf R}^\prime)d\mbox{\it\bf r} \\& = & \sum_{\mbox{\it\bf R}}\e^{-i\mbox{\it\bf k}\cdot\mbox{\it\bf R}} \int \psi^{a*}_n(\mbox{\it\bf r}-\mbox{\it\bf R})\delta{\cal V}(\mbox{\it\bf r})\psi^a_n(\mbox{\it\bf r})d\mbox{\it\bf r} \end{eqnarray},$} dove al primo passaggio si č sostituito {$\mbox{\it\bf R}\rightarrow\mbox{\it\bf R}+\mbox{\it\bf R}^\prime$} nella prima delle due sommatorie sul reticolo e si č sfruttata la periodicitŕ di {$\delta{\cal V}$}, al secondo passaggio si č sostituita la variabile d'integrazione {$\mbox{\it\bf r}\rightarrow\mbox{\it\bf r}-\mbox{\it\bf R}^\prime$}, e da ultimo la sommatoria su {$\mbox{\it\bf R}^\prime$} di termini indipendenti da {$\mbox{\it\bf R}^\prime$} ha fornito un fattore {$N$}. Ora dobbiamo valutare quali termini di queste somme danno contributi apprezzabili, ricordando che le funzioni d'onda atomiche decadono rapidamente. Le loro code si estendono ai primi vicini, ed il fattore {$\delta{\cal V}={\cal V} - \sum_{\mbox{\it\bf R}}{\cal V}^a(\mbox{\it\bf r}+\mbox{\it\bf R})$}, che contiene il potenziale periodico, intenso su tutti i siti, ne amplifica il contributo agli integrali della 31; viceversa per semplicitŕ supporremo che la sovrapposizione tra due funzioni d'onda centrate su siti secondi vicini sia sufficientemente piccola da rendere trascurabili i corrispondenti integrali (l'approssimazione naturalmente migliora includendo qualche termine in piů). In definitiva possiamo scrivere {$(32)\qquad\qquad \begin{eqnarray}\varepsilon_n(\mbox{\it\bf k})-\varepsilon^a_n & \approx & \int \left|\psi^a_n(\mbox{\it\bf r})\right|^2\delta{\cal V}(\mbox{\it\bf r})d\mbox{\it\bf r} + \sum_{\mbox{\it\bf R} } \e^{i\mbox{\it\bf k}\cdot\mbox{\it\bf R}} \int \psi^{a*}_n(\mbox{\it\bf r}-\mbox{\it\bf R})\delta{\cal V}(\mbox{\it\bf r})\psi^{a}_n(\mbox{\it\bf r})d\mbox{\it\bf r} \\ & \equiv & - \alpha_n + \sum_{\mbox{\it\bf R} } \gamma_{n\mbox{\it\bf R}} e^{i\mbox{\it\bf k}\cdot\mbox{\it\bf R}}\end{eqnarray} ,$} dove si č definito {$\alpha_n\equiv\int \left|\psi^a_n(\mbox{\it\bf r})\right|^2\delta{\cal V}(\mbox{\it\bf r})d\mbox{\it\bf r}$} (una quantitŕ intrinsecamente positiva, visto che il cristallo č stabile), {$\gamma_{n\mbox{\it\bf R}} \equiv \int \psi^{a*}_n(\mbox{\it\bf r}-\mbox{\it\bf R}) \delta{\cal V}(\mbox{\it\bf r}) \psi^{a}_n(\mbox{\it\bf r}) d\mbox{\it\bf r}$}, e la somma su {$\mbox{\it\bf R}$} č limitata ai vettori di traslazione che congiungono i primi vicini all'origine. [1] Il modello conserva la sua validitŕ finchč {$\gamma_{\mbox{\it\bf a}}$} resta una quantitŕ piccola rispetto alla separazione tra i livelli atomici.
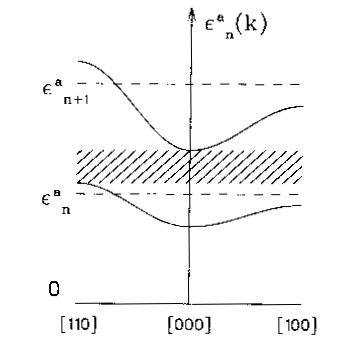
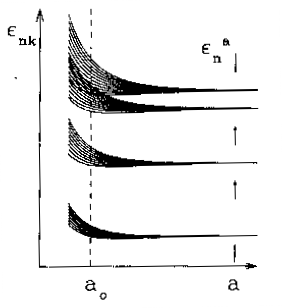
Gli intervalli proibiti originano direttamente dalla separazione tra gli autovalori discreti dell'energia dell'atomo componente; nel solido l'interazione col potenziale periodico (attraverso {$\gamma_{n\mbox{\it\bf R}}$}) apre ciascun autovalore in una banda quasi continua, composta da un fitto insieme di {$N\sim 10^{23}$} livelli energetici, come mostra schematicamente la figura 25.
< Condizioni al contorno di Born-Von Karman e numero degli stati | Indice | Bande e classificazione dei solidi > |