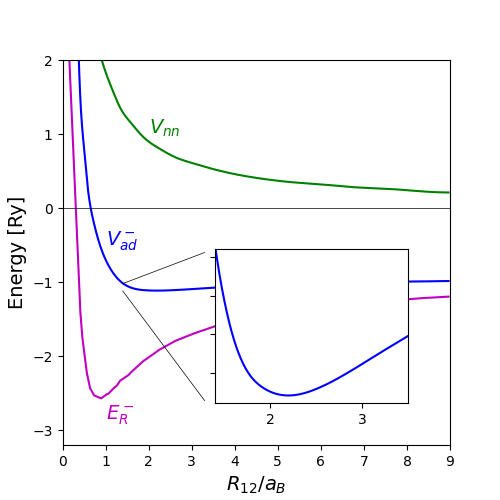
|
L'equazione di Schr÷dinger di molecole e solidi si
scrive a partire da una Hamiltoniana formalmente identica a quella di una
molecola. La scriviamo qui per comoditÓ nel caso
monoatomico, separando i
termini elettronici da quelli che contengono solo operatori nucleari
{$$\begin{align} \tag{1} {\cal H} &= \sum_{i=1}^n \left({{p_i^2}\over{2m}} - \sum_{l=1}^{N} { {Ze^2} \over {|\mathbf{r}_i-\mathbf{R}_l}| }\right) + \frac 1 2 \sum_{i\not=j=1}^n { {e^2} \over {|\mathbf{r}_i-\mathbf{ r}_j|} } \\ & \quad + \sum_{l=1}^{N} { {P_{l}^2} \over {2M} } + {1\over 2} \sum_{l\not=m=1}^{N} { {Z^2e^2} \over {|\mathbf{ R}_l-\mathbf{R}_m}| }, \end{align}$$}
dove {$\mathbf{p}_i=-i\hbar\boldsymbol\nabla_{\mathbf{r}_i}$} e {$\mathbf{r}_i$} sono gli operatori
momento e posizione degli {$n$} elettroni, mentre
{$\mathbf{P}_l=-i\hbar\boldsymbol\nabla_{\mathbf R_l}$} ed {${\mathbf R}_l$} sono i corrispondenti operatori
per gli {$N$} nuclei.
Riscriviamo l'equazione totale agli stati stazionari in maniera simbolica, indicando com {$R$}, ($r$) le variabili spaziali (vettoriali) nucleari (elettroniche), con {$K$} i termini cinetici, {$V = V(r-R), V_e = V_e(r), V_n = V_n(R)$} rispettivamente i potenziali elettroni-nuclei, elettroni-elettroni e nuclei-nuclei. La discussione che segue Þ sostanzialmente corretta, ma cerca di evitare le complicazioni della notazione pedante dell'Hamiltoniana (1). L'equazione di Schr÷dinger si riscrive quindi:
{$$ \tag{2} {\cal H}\, \phi(R)\psi_R(r) = (K_R + \underbrace{K_r + V + V_e}_{{\cal H}_e} + V_n) \,\phi(R)\psi_R(r) = E \phi(R)\,\psi_R(r)$$}
dove abbiamo assunto che la funzione d'onda totale sia fattorizzabile in nucleare e elettronica,{$\Psi(R,r)= \phi(R)\psi_R(r)$}, e la funzione d'onda elettronica {$\psi$}, che dipende parametricamente da {$R$} obbedisce all'equazione di Schroedinger
{$$ \tag{3} {\cal H}_e \psi^\alpha_R = E^\alpha_R \psi_R^\alpha\qquad \alpha=0,1,\cdot$$}
per gli {$\alpha$} autostati e autovalori. La fattorizzazione vale solo se si riesce a separare anche un termine dell'Hamiltoniana che rappresenta solo i nuclei, {${\cal H}_n$} e non esistono termini di interazione. In realtÓ ci sono due termini di interazione tra elettroni e nuclei. {$V$}, che abbiamo inglobato in {${\cal H}_e$} e che dÓ una dipendenza da {$R$} a {$\psi_R$}. Per il momento l'abbiamo definita parametrica, dovremo chiarire meglio perchÞ. E il termine {$K_R$} il cui Laplaciano nucleare agisce anche sulla dipendenza parametrica della funzione d'onda elettronica, dipendenz che supponiamo corrispondere ad una funzione analitica di {$R$}. Ossia
{$$ K_R \phi(R)\psi_R(r) = -\frac {\hbar^2 \nabla^2_R}{2M} \phi\psi = \psi K_R \phi+ \phi K_R \psi - 2 \frac {\hbar^2}{2M} \boldsymbol{\nabla}_R \phi\cdot\boldsymbol{\nabla}_R\psi $$}
|